Analysis Module API¶
Module for analyzing MD trajectories and NAMD energy data with easy-to-use wrapper classes supporting all GUI options.
Overview¶
The analysis module provides three main trajectory/energy classes:
EnergyAnalyzer- Parse and plot NAMD energy data with full customizationTrajectoryAnalyzer- Calculate and plot RMSD, RMSF, distances, and radius of gyration with complete controlBilayerTrajectoryAnalyzer- Calculate area per lipid and membrane thickness using lipyphilic
Key Features:
- Simple 2-line API for quick analysis
- Full customization with all GUI options
- Multi-file support with proper time scaling
- Unit conversions (Å/nm, ps/ns/µs, kcal/kJ)
- Publication-quality plots (300 DPI)
Import¶
from gatewizard.utils.namd_analysis import (
EnergyAnalyzer,
TrajectoryAnalyzer,
BilayerTrajectoryAnalyzer,
run_bilayer_analysis,
get_equilibration_progress
)
Using Custom Time Input in the GUI¶
For Trajectory Files (.dcd, .xtc, .trr)¶
When analyzing multiple trajectory files in the GUI Structural Analysis tab:
- Add Files: Click "Add Files" or "Add Folder" to load trajectory files
- Input Time: Each file will appear in the list with a time input field showing "Time (ns)"
- Enter Duration: Enter the simulation duration for each file in nanoseconds
- Example: For a 50 ps simulation, enter
0.05(50 ps = 0.05 ns) - Auto-Detect: Click "Auto-Detect Time" to automatically extract times from DCD headers
- Reorder: Drag files using the
:::handle to reorder if needed
Example File List:
::: step1_equilibration.dcd [0.05] ns
::: step2_equilibration.dcd [0.05] ns
::: step3_equilibration.dcd [0.05] ns
For Energy Log Files (.log)¶
When analyzing NAMD log files in the GUI Energetic Analysis tab:
- Add Files: Click "Add Files" or "Add Folder" to load NAMD log files
- Input Time: Each file will appear with a time input field
- Enter Duration: Enter the simulation duration for each file in nanoseconds
- Example: For step1_equilibration.log (50 ps), enter
0.05 - Auto-Detect: Click "Auto-Detect Time" to extract from log file timestamps
- Plot: The time axis will scale correctly across all files
Example File List:
::: step1_equilibration.log [0.05] ns
::: step2_equilibration.log [0.05] ns
::: step7_production.log [0.10] ns
Important Notes:
- Time input is the duration of each file, not cumulative
- Files are processed in order - time is sequential (file1: 0-50ps, file2: 50-100ps, etc.)
- Leave empty or 0 to use default timestep-based calculation
- Time unit is always nanoseconds in the input fields
- You can change display units (ps/ns/µs) in Plot Settings
Class: EnergyAnalyzer¶
Easy-to-use wrapper for NAMD energy analysis with built-in plotting and customization.
Constructor¶
EnergyAnalyzer(
log_file: Union[Path, str, List[Union[Path, str]]],
file_times: Optional[Dict[str, float]] = None
)
Initialize energy analyzer with NAMD log file(s).
Parameters:
log_file (Path, str, or List): Path to NAMD log file, or list of paths for multi-file analysis
- Single file:
"step1_equilibration.log" - Multiple files:
["step1.log", "step2.log", "step3.log"]
file_times (Dict[str, float], optional): Mapping of log filenames to their simulation durations in nanoseconds
- Example:
{"step1.log": 0.05, "step2.log": 0.05, "step3.log": 0.05} - Important: Use just the filename (not full path) as the key
- Time is the duration of each file, not cumulative
- Note: In the GUI, you can input times for each file directly in the file list
Examples:
# Single file
analyzer = EnergyAnalyzer("step1_equilibration.log")
# Multiple files with custom time
analyzer = EnergyAnalyzer(
["step1.log", "step2.log", "step3.log"],
file_times={
"step1.log": 0.05, # 50 ps = 0.05 ns
"step2.log": 0.05,
"step3.log": 0.05
}
)
Method: plot_energy()¶
Create a 4-panel energy analysis plot with full customization.
plot_energy(
energy_units: str = "kcal/mol",
time_units: str = "ns",
target_temperature: Optional[float] = None,
target_pressure: Optional[float] = None,
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
title: Optional[str] = None,
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (12, 10),
dpi: int = 300
)
Parameters:
| Parameter | Type | Default | Description |
|---|---|---|---|
energy_units | str | "kcal/mol" | Energy units: "kcal/mol" or "kJ/mol" |
time_units | str | "ns" | Time units: "ps", "ns", or "µs" |
target_temperature | float or None | None | Target temperature for reference line (K). Defaults to 300.0 K if not provided. |
target_pressure | float or None | None | Target pressure for reference line (atm). Defaults to 1.0 atm if not provided. |
bg_color | str | "#2b2b2b" | Plot area background color (hex or "none") |
fig_bg_color | str | "#212121" | Figure border background color (hex or "none") |
text_color | str | "Auto" | Text/axes color ("Auto", color name, or hex) |
show_grid | bool | True | Show grid lines on plots |
title | str or None | None | Custom main title (auto-generated if None) |
save | str or None | None | Filename to save plot |
show | bool | False | Display plot interactively |
figsize | tuple | (12, 10) | Figure size (width, height) in inches |
dpi | int | 300 | Resolution for saved figure |
Plots Generated:
- Total Energy - Convergence over time
- Potential & Kinetic - Energy components
- Temperature - Stability with target line
- Pressure - Fluctuations (or energy components if no pressure)
Method: plot_properties()¶
Create custom plots of selected energy/thermodynamic properties with full control over visualization.
This method provides complete API access to all GUI plotting capabilities, allowing users to:
- Analyze multiple log files with custom time assignments
- Select specific properties to plot
- Combine or separate data from multiple files
- Apply full customization (colors, units, grid, limits, etc.)
plot_properties(
properties: Union[str, List[str]],
separate_plots: bool = False,
energy_units: str = "kcal/mol",
time_units: str = "ns",
line_colors: Optional[Union[str, List[str]]] = None,
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
grid_color: str = "#444444",
show_grid: bool = True,
xlim: Optional[tuple] = None,
ylim: Optional[tuple] = None,
title: Optional[str] = None,
save_prefix: Optional[str] = None,
show: bool = False,
figsize: tuple = (10, 6),
dpi: int = 300
)
Parameters:
| Parameter | Type | Default | Description |
|---|---|---|---|
properties | str or List[str] | - | Property name(s) to plot. See available properties below. |
separate_plots | bool | False | If True, create individual plots per file; if False, combine all files on same plot |
energy_units | str | "kcal/mol" | Energy units: "kcal/mol" or "kJ/mol" |
time_units | str | "ns" | Time units: "ps", "ns", or "µs" |
line_colors | str or List[str] | None | Line color(s). Single color or list for multiple files. Auto-assigned if None. |
bg_color | str | "#2b2b2b" | Plot area background color (hex, color name, or "none") |
fig_bg_color | str | "#212121" | Figure border background color (hex, color name, or "none") |
text_color | str | "Auto" | Text/axes color. "Auto" auto-detects from bg luminance. |
grid_color | str | "#444444" | Grid line color (hex or color name) |
show_grid | bool | True | Show grid lines |
xlim | tuple or None | None | X-axis limits: (min, max) |
ylim | tuple or None | None | Y-axis limits: (min, max) |
title | str or None | None | Custom plot title (auto-generated if None) |
save_prefix | str or None | None | Prefix for saved filenames. Files saved as {prefix}_{property}.png |
show | bool | False | Display plot interactively |
figsize | tuple | (10, 6) | Figure size (width, height) in inches |
dpi | int | 300 | Resolution for saved figures |
Available Properties:
Use get_available_properties() method to see all properties in your log file(s).
Note: Property names are case-insensitive and support multiple formats. For example, to plot temperature, you can use:
"TEMP","temp","Temp"(short names)"Temperature","TEMPERATURE","temperature"(full names)- Any mixed case variation like
"TeMpErAtUrE"will also work!
The same applies to all properties - use whatever format is most convenient for you.
| Property | Alternative Names | Description | Units |
|---|---|---|---|
"TOTAL" | "Total", "total", "Total Energy", etc. | Total energy | kcal/mol or kJ/mol |
"KINETIC" | "Kinetic", "kinetic", "kin", "Kinetic Energy" | Kinetic energy | kcal/mol or kJ/mol |
"POTENTIAL" | "Potential", "potential", "pot", "Potential Energy" | Potential energy | kcal/mol or kJ/mol |
"ELECT" | "Elect", "elect", "elec", "Electrostatic Energy" | Electrostatic energy | kcal/mol or kJ/mol |
"VDW" | "vdw", "Vdw", "Van der Waals Energy" | van der Waals energy | kcal/mol or kJ/mol |
"TEMP" | "temp", "Temp", "Temperature" | Temperature | K |
"PRESSURE" | "pressure", "Pressure", "press", "Press" | Pressure | bar |
"VOLUME" | "volume", "Volume", "vol", "Vol" | System volume | Ų |
"BOND" | "bond", "Bond", "Bond Energy" | Bond energy | kcal/mol or kJ/mol |
"ANGLE" | "angle", "Angle", "Angle Energy" | Angle energy | kcal/mol or kJ/mol |
"DIHEDRAL" | "dihedral", "Dihedral", "Dihedral Energy" | Dihedral energy | kcal/mol or kJ/mol |
"IMPROPER" | "improper", "Improper", "Improper Energy" | Improper energy | kcal/mol or kJ/mol |
Example 1: Basic Energy Analysis¶
Simplest energy analysis using plot_energy() - 4-panel plot with default settings.
from pathlib import Path
from gatewizard.utils.namd_analysis import EnergyAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Multiple log files from equilibration_folder
log_files = [
data_dir / "step1_equilibration.log",
data_dir / "step2_equilibration.log",
data_dir / "step3_equilibration.log"
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = EnergyAnalyzer(
[str(f) for f in log_files],
file_times={
"step1_equilibration.log": 0.1, # 100 ps
"step2_equilibration.log": 0.1, # 100 ps
"step3_equilibration.log": 0.1 # 100 ps
}
)
# Generate comprehensive 4-panel energy plot
analyzer.plot_energy(target_temperature=300, # 300 K
target_pressure=1.01325, # 1.01325 atm (1 bar)
time_units="ps",
save="energy_analysis_example_01.png")
print(f"Energy analysis complete!")
print(f"Plot saved: energy_analysis_example_01.png")
Output figure:
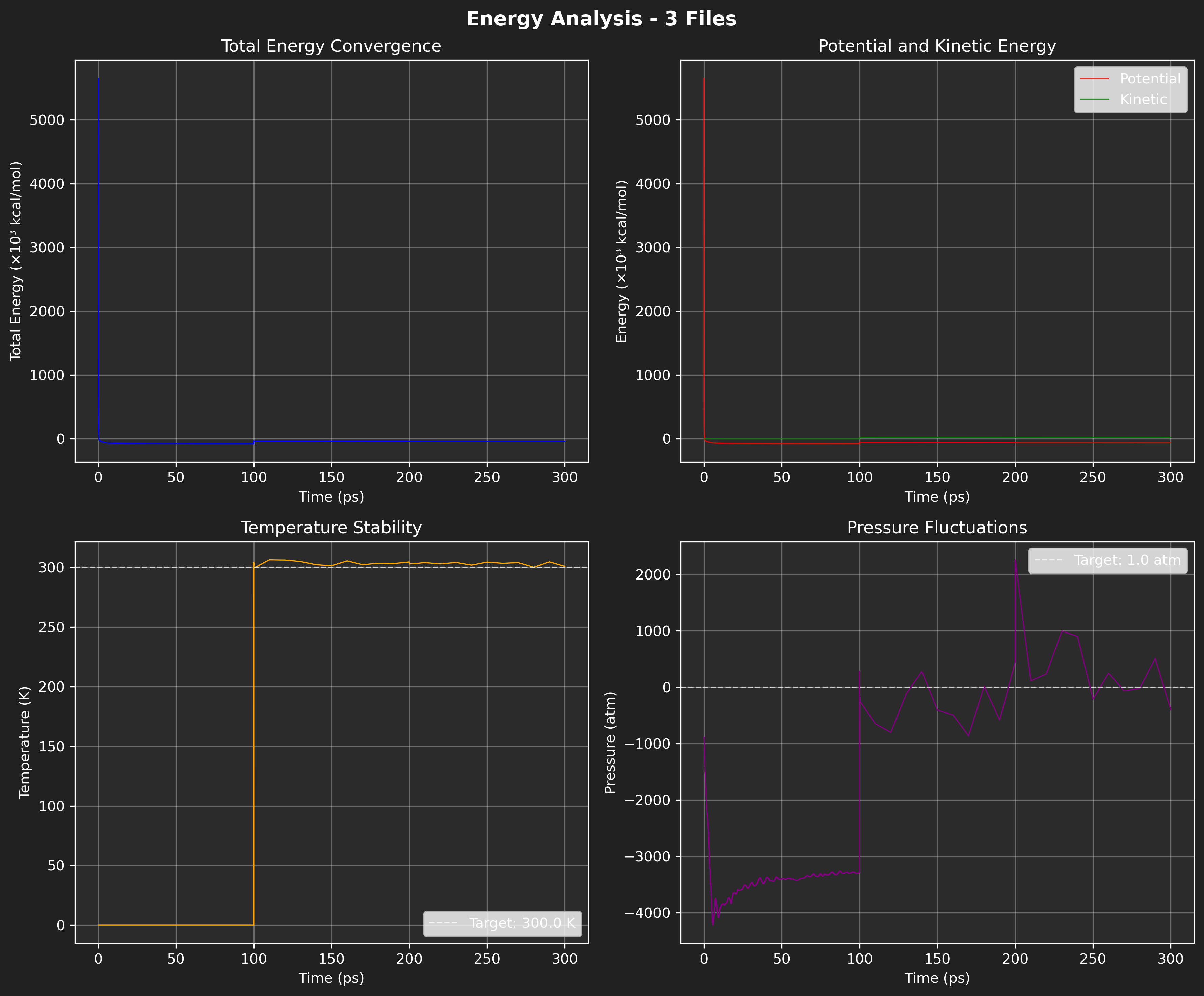
Figure: Default energy plot using the specified NAMD .log files.
Multi-File Color Assignment:
When plotting multiple files on the same plot, colors are automatically assigned:
- If
line_colorsis a single color: all files use that color - If
line_colorsis a list: each file gets corresponding color - If
line_colors=None: auto-assigned from matplotlib color cycle
Time Scaling with file_times:
The file_times parameter in the constructor allows mapping data points to correct time ranges:
# Example: 3 files with different simulation times
# File 1: 0-50 ns (5000 frames)
# File 2: 50-125 ns (7500 frames)
# File 3: 125-200 ns (7500 frames)
analyzer = EnergyAnalyzer(
log_file=["file1.log", "file2.log", "file3.log"],
file_times=[(0, 50), (50, 125), (125, 200)],
# or
#file_times={
# "file1.log": 0.05, # 50 ps = 0.05 ns
# "file2.log": 0.05,
# "file3.log": 0.05
#}
)
# Now time axis will correctly show 0-200 ns with proper frame distribution
analyzer.plot_properties(properties="TEMP", time_units="ns")
Method: get_available_properties()¶
Get list of all plottable properties available in the loaded log file(s).
Returns: List of property names that can be passed to plot_properties()
Example 2: Get available properties¶
from pathlib import Path
from gatewizard.utils.namd_analysis import EnergyAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
# Path to log file in equilibration_folder
log_file = script_dir / "equilibration_folder" / "step1_equilibration.log"
# Initialize energy analyzer
analyzer = EnergyAnalyzer(log_file)
props = analyzer.get_available_properties()
print(f"Can plot: {', '.join(props)}")
# Output: Can plot: Total Energy, Potential Energy, Kinetic Energy,
# Electrostatic Energy, Van der Waals Energy, Bond Energy,
# Angle Energy, Dihedral Energy, Improper Energy, Pressure, Volume
Example 3: Individual energy plots¶
from pathlib import Path
from gatewizard.utils.namd_analysis import EnergyAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Multiple log files from equilibration_folder
log_files = [
data_dir / "step1_equilibration.log",
data_dir / "step2_equilibration.log",
data_dir / "step3_equilibration.log",
data_dir / "step4_equilibration.log",
data_dir / "step5_equilibration.log",
data_dir / "step6_equilibration.log",
data_dir / "step7_production.log"
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = EnergyAnalyzer(
[str(f) for f in log_files],
file_times={
"step1_equilibration.log": 0.1, # 100 ps
"step2_equilibration.log": 0.1, # 100 ps
"step3_equilibration.log": 0.1, # 100 ps
"step4_equilibration.log": 0.1, # 100 ps
"step5_equilibration.log": 0.1, # 100 ps
"step6_equilibration.log": 0.1, # 100 ps
"step7_production.log": 0.1, # 100 ps
}
)
# Plot specific energy properties
analyzer.plot_properties(
properties=["bond energy", "angle energy", "dihedral energy"],
energy_units="kcal/mol",
time_units="ps",
bg_color = "#ffffff",
fig_bg_color = "#FFFFFF",
save="energy_analysis_example_03.png",
dpi=300,
)
print(f"Energy properties plot complete!")
print(f"Plot saved: energy_analysis_example_03.png")
Example Output:
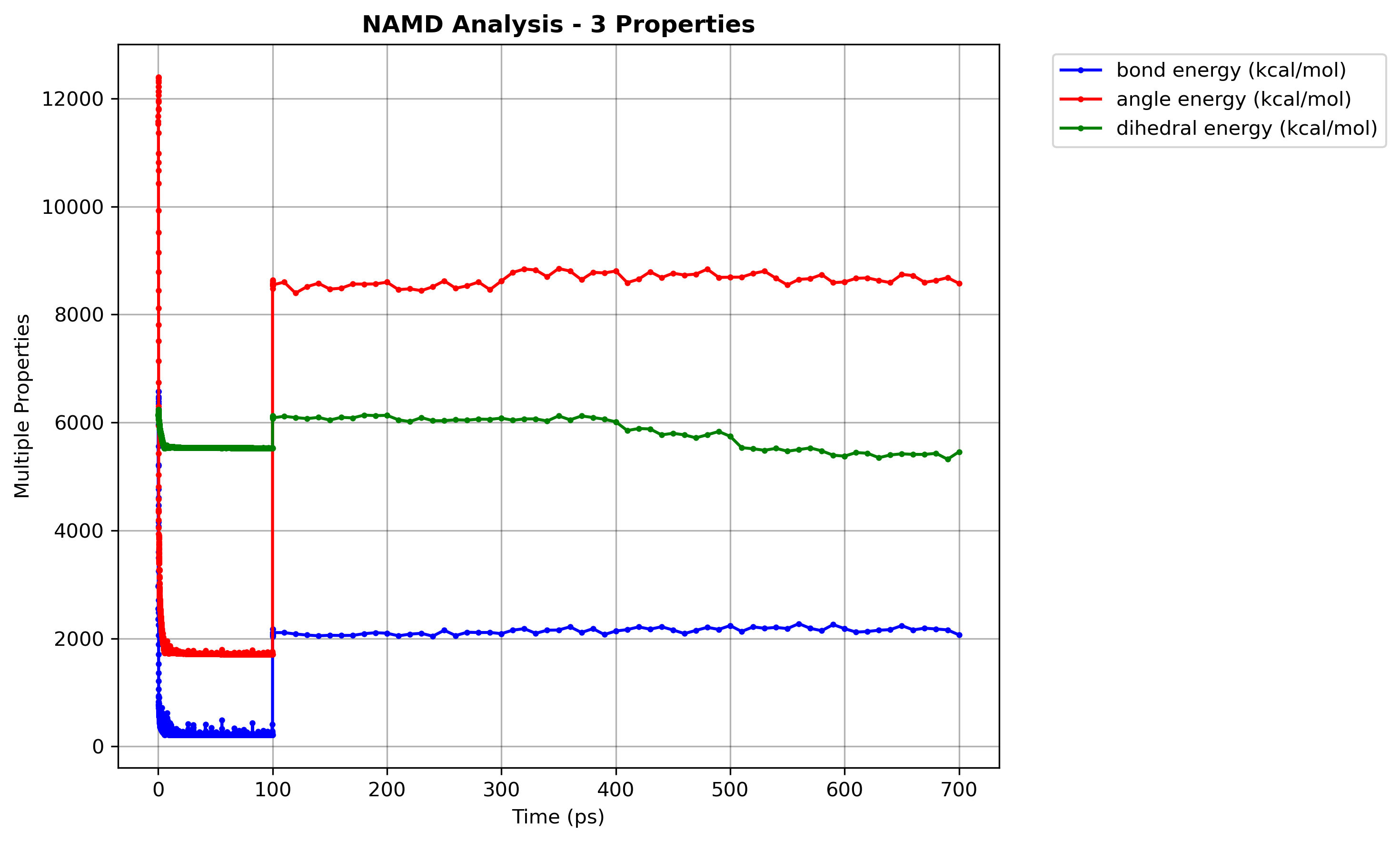
Figure: Multiple energy properties plot.
Method: get_statistics()¶
Get statistical summary of all energy components.
Returns: Dictionary with statistics for each energy term
Statistics Provided:
mean- Average valuestd- Standard deviationmin- Minimum valuemax- Maximum valueinitial- First valuefinal- Last value
Available Energy Terms:
timestep,total,kinetic,potentialelect,vdw,boundary,misctemp,pressure,volume
Example 4: Individual energy plots¶
from pathlib import Path
from gatewizard.utils.namd_analysis import EnergyAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
# Path to log file in equilibration_folder
log_file = script_dir / "equilibration_folder" / "step2_equilibration.log"
# Initialize energy analyzer
analyzer = EnergyAnalyzer(log_file)
stats = analyzer.get_statistics()
# Temperature
temp = stats['temp']
print(f"Temperature: {temp['mean']:.1f} ± {temp['std']:.1f} K")
print(f" Range: {temp['min']:.1f} - {temp['max']:.1f} K")
# Energy
energy = stats['bond']
print(f"Bond Energy: {energy['mean']:.0f} kcal/mol")
print(f" Final: {energy['final']:.0f} kcal/mol")
print(f" Convergence: {abs(energy['final'] - energy['mean']):.0f} kcal/mol")
# Output:
# Temperature: 303.6 ± 2.1 K
# Range: 299.4 - 306.3 K
# Bond Energy: 2080 kcal/mol
# Final: 2097 kcal/mol
# Convergence: 17 kcal/mol
Class: TrajectoryAnalyzer¶
Easy-to-use wrapper for MD trajectory analysis with built-in plotting and full customization.
Constructor¶
TrajectoryAnalyzer(
topology: Path,
trajectory: Path | List[Path],
file_times: Optional[Dict[str, float]] = None
)
Initialize trajectory analyzer with topology and trajectory file(s).
Parameters:
topology(Path or str): Path to topology file (PSF, PDB, IMPCRD, etc.)trajectory(Path, str, or List): Path(s) to trajectory file(s) (DCD, XTC, TRR, etc.)- Single file:
"trajectory.dcd" - Multiple files:
["eq1.dcd", "eq2.dcd", "prod.dcd"] file_times(Dict[str, float], optional): Mapping of trajectory filenames to their simulation durations in nanoseconds
Example: {"eq1.dcd": 0.05, "eq2.dcd": 0.05, "prod.dcd": 0.05}
- Important: Use just the filename (not full path) as the key
- Time is the duration of that file, not cumulative
- Note: In the GUI, you can input times for each file directly in the file list
Examples:
Single Trajectory:
Multiple Trajectories with Time Scaling:
analyzer = TrajectoryAnalyzer(
"system.pdb",
["step1_equilibration.dcd", "step2_equilibration.dcd", "step7_production.dcd"],
file_times={
"step1_equilibration.dcd": 0.05, # 50 ps
"step2_equilibration.dcd": 0.05, # 50 ps
"step7_production.dcd": 0.05 # 50 ps
}
)
# Time axis will be continuous: [0...0.05] [0.05...0.10] [0.10...0.15] ns
Method: calculate_rmsd()¶
Calculate RMSD for selected atoms.
calculate_rmsd(
selection: str = "protein and backbone",
reference_frame: int = 0,
align: bool = True
) -> Dict[str, np.ndarray]
Parameters:
selection(str): MDTraj selection stringreference_frame(int): Frame to use as reference (0 = first frame)align(bool): If True, perform structural alignment (rotation + translation) before RMSD. If False, calculate raw coordinate RMSD without alignment.
Returns: Dictionary with:
'time': Time array in nanoseconds'rmsd': RMSD values in Angstroms
Example:
data = analyzer.calculate_rmsd("protein and backbone", align=True)
print(f"Time range: {data['time'][0]:.3f} - {data['time'][-1]:.3f} ns")
print(f"RMSD range: {data['rmsd'].min():.2f} - {data['rmsd'].max():.2f} Å")
Method: plot_rmsd()¶
Plot RMSD with full customization options including line styling, threshold highlighting, and convergence line control.
plot_rmsd(
selection: str = "protein and backbone",
reference_frame: int = 0,
align: bool = True,
distance_units: str = "Å",
time_units: str = "ns",
line_color: str = "blue",
line_width: float = 1.2,
line_style: str = "-",
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
xlim: Optional[tuple] = None,
ylim: Optional[tuple] = None,
title: Optional[str] = None,
xlabel: Optional[str] = None,
ylabel: Optional[str] = None,
highlight_threshold: Optional[float] = None,
highlight_color: str = "orange",
highlight_alpha: float = 0.2,
show_convergence: bool = True,
convergence_color: str = "red",
convergence_style: str = "--",
convergence_width: float = 1.5,
hlines: Optional[List[float]] = None,
hline_colors: Optional[List[str]] = None,
hline_styles: Optional[List[str]] = None,
hline_widths: Optional[List[float]] = None,
vlines: Optional[List[float]] = None,
vline_colors: Optional[List[str]] = None,
vline_styles: Optional[List[str]] = None,
vline_widths: Optional[List[float]] = None,
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (10, 6),
dpi: int = 300
)
Parameters:
| Parameter | Type | Default | Description |
|---|---|---|---|
selection | str | "protein and backbone" | MDTraj selection string |
reference_frame | int | 0 | Reference frame index |
align | bool | True | Perform structural alignment before RMSD |
distance_units | str | "Å" | Distance units: "Å" or "nm" |
time_units | str | "ns" | Time units: "ps", "ns", or "µs" |
line_color | str | "blue" | Plot line color (matplotlib color or hex) |
line_width | float | 1.2 | Line thickness/width |
line_style | str | "-" | Line style: "-" (solid), "--" (dashed), "-." (dash-dot), ":" (dotted) |
bg_color | str | "#2b2b2b" | Plot area background (hex or "none") |
fig_bg_color | str | "#212121" | Figure border background (hex or "none") |
text_color | str | "Auto" | Text/axes color ("Auto", color, or hex) |
show_grid | bool | True | Show grid lines |
xlim | tuple or None | None | X-axis limits (min, max) |
ylim | tuple or None | None | Y-axis limits (min, max) |
title | str or None | None | Custom title (auto-generated if None) |
xlabel | str or None | None | Custom X-label (auto with units if None) |
ylabel | str or None | None | Custom Y-label (auto with units if None) |
highlight_threshold | float or None | None | Highlight regions above this RMSD value |
highlight_color | str | "orange" | Color for highlight region and line |
highlight_alpha | float | 0.2 | Alpha transparency for highlight fill |
show_convergence | bool | True | Show convergence line (mean of last 20% of trajectory) |
convergence_color | str | "red" | Color for convergence line |
convergence_style | str | "--" | Line style for convergence line |
convergence_width | float | 1.5 | Width of convergence line |
hlines | list or None | None | List of Y values for horizontal reference lines |
hline_colors | list or None | None | List of colors for horizontal lines |
hline_styles | list or None | None | List of line styles for horizontal lines (default: "--") |
hline_widths | list or None | None | List of line widths for horizontal lines (default: 1.0) |
vlines | list or None | None | List of X values for vertical reference lines |
vline_colors | list or None | None | List of colors for vertical lines |
vline_styles | list or None | None | List of line styles for vertical lines (default: "--") |
vline_widths | list or None | None | List of line widths for vertical lines (default: 1.0) |
save | str or None | None | Filename to save plot |
show | bool | False | Display plot interactively |
figsize | tuple | (10, 6) | Figure size (width, height) |
dpi | int | 300 | Resolution for saved figure |
Example 5: RMSD Plot¶
from pathlib import Path
from gatewizard.utils.namd_analysis import TrajectoryAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Path to topology in equilibration_folder
topology_file = data_dir / "system.pdb"
# Multiple trajectory files from equilibration_folder
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd"
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = TrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times={
"step1_equilibration.dcd": 0.1, # 100 ps
"step2_equilibration.dcd": 0.1, # 100 ps
"step3_equilibration.dcd": 0.1 # 100 ps
}
)
# Plot RMSD across all trajectories
analyzer.plot_rmsd(
selection="protein and backbone",
time_units="ns",
bg_color="white",
fig_bg_color="white",
text_color="black",
show_grid=False,
line_color="#1f77b4",
line_width=2,
#title=" ",
save="trajectory_analysis_example_05.png",
dpi=300,
# other settings...
)
print(f"Multi-file trajectory analysis complete!")
print(f"Plot saved: trajectory_analysis_example_05.png")
print(f"Total simulation time: 300 ps")
Method: calculate_rmsf()¶
Calculate RMSF (Root Mean Square Fluctuation) for selected atoms.
Parameters:
selection(str): MDTraj selection string
Returns: Dictionary with:
'resids': Residue numbers'rmsf': RMSF values in Angstroms'resnames': Residue names (e.g., ALA, GLY, VAL)'atom_indices': Atom indices
Example 6: RMSF Calculation¶
from pathlib import Path
from gatewizard.utils.namd_analysis import TrajectoryAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Path to topology in equilibration_folder
topology_file = data_dir / "system.pdb"
# Multiple trajectory files from equilibration_folder
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd"
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = TrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times={
"step1_equilibration.dcd": 0.1, # 100 ps
"step2_equilibration.dcd": 0.1, # 100 ps
"step3_equilibration.dcd": 0.1 # 100 ps
}
)
# Calculate RMSF for alpha carbon atoms
data = analyzer.calculate_rmsf("protein and name CA")
print(f"Residues analyzed: {len(data['resids'])}")
print(f"RMSF range: {data['rmsf'].min():.2f} - {data['rmsf'].max():.2f} Å")
print(f"Mean RMSF: {data['rmsf'].mean():.2f} Å")
# Output:
# Residues analyzed: 26
# RMSF range: 0.09 - 0.15 Å
# Mean RMSF: 0.12 Å
Method: plot_rmsf()¶
Plot RMSF with full customization including line styling, residue labeling, and flexibility highlighting.
plot_rmsf(
selection: str = "protein and name CA",
xaxis_type: str = "residue_number",
show_residue_labels: bool = True,
residue_name_format: str = "single",
label_frequency: str = "auto",
distance_units: str = "Å",
line_color: str = "blue",
line_width: float = 1.2,
line_style: str = "-",
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
xlim: Optional[tuple] = None,
ylim: Optional[tuple] = None,
title: Optional[str] = None,
xlabel: Optional[str] = None,
ylabel: Optional[str] = None,
highlight_threshold: Optional[float] = None,
highlight_color: str = "orange",
highlight_alpha: float = 0.2,
hlines: Optional[List[float]] = None,
hline_colors: Optional[List[str]] = None,
hline_styles: Optional[List[str]] = None,
hline_widths: Optional[List[float]] = None,
vlines: Optional[List[float]] = None,
vline_colors: Optional[List[str]] = None,
vline_styles: Optional[List[str]] = None,
vline_widths: Optional[List[float]] = None,
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (12, 6),
dpi: int = 300
)
Parameters:
| Parameter | Type | Default | Description |
|---|---|---|---|
selection | str | "protein and name CA" | MDTraj selection string |
xaxis_type | str | "residue_number" | X-axis type: "residue_number", "residue_type_number", "atom_index" |
show_residue_labels | bool | True | Show residue labels on X-axis |
residue_name_format | str | "single" | Residue format: "single" (A, G, V) or "triple" (ALA, GLY, VAL) |
label_frequency | str | "auto" | Label frequency: "all", "auto", "every_2", "every_5", "every_10", "every_20" |
distance_units | str | "Å" | Distance units: "Å" or "nm" |
line_color | str | "blue" | Plot line color (matplotlib color or hex) |
line_width | float | 1.2 | Line thickness/width |
line_style | str | "-" | Line style: "-" (solid), "--" (dashed), "-." (dash-dot), ":" (dotted) |
highlight_threshold | float or None | None | If set, highlight residues above this RMSF value |
highlight_color | str | "orange" | Color for highlight region and threshold line |
highlight_alpha | float | 0.2 | Alpha transparency for highlight fill region |
hlines | list or None | None | List of Y values for horizontal reference lines |
hline_colors | list or None | None | List of colors for horizontal lines (cycles if fewer than lines) |
hline_styles | list or None | None | List of line styles for horizontal lines (default: "--") |
hline_widths | list or None | None | List of line widths for horizontal lines (default: 1.0) |
vlines | list or None | None | List of X values (residue numbers) for vertical reference lines |
vline_colors | list or None | None | List of colors for vertical lines (cycles if fewer than lines) |
vline_styles | list or None | None | List of line styles for vertical lines (default: "--") |
vline_widths | list or None | None | List of line widths for vertical lines (default: 1.0) |
| ...other params | Same as plot_rmsd() for colors, grid, limits, etc. |
X-Axis Types:
"residue_number"- Simple numbers: 1, 2, 3, ..."residue_type_number"- Type + number: ALA1, GLY2, VAL3, ..."atom_index"- Atom indices: 0, 15, 30, ...
Label Frequencies:
"all"- Every residue labeled"auto"- Smart frequency based on count (<20: all, <50: every_2, <100: every_5, <200: every_10, else: every_20)"every_2","every_5","every_10","every_20"- Specific intervals
Example 7: RMSF Plotting¶
from pathlib import Path
from gatewizard.utils.namd_analysis import TrajectoryAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Path to topology in equilibration_folder
topology_file = data_dir / "system.pdb"
# Multiple trajectory files from equilibration_folder
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd",
data_dir / "step4_equilibration.dcd",
data_dir / "step5_equilibration.dcd",
data_dir / "step6_equilibration.dcd",
data_dir / "step7_production.dcd",
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = TrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times={
"step1_equilibration.dcd": 0.1, # 100 ps
"step2_equilibration.dcd": 0.1, # 100 ps
"step3_equilibration.dcd": 0.1, # 100 ps
"step4_equilibration.dcd": 0.1, # 100 ps
"step5_equilibration.dcd": 0.1, # 100 ps
"step6_equilibration.dcd": 0.1, # 100 ps
"step7_production.dcd": 0.1 # 100 ps
}
)
# Plot RMSF across all trajectories
analyzer.plot_rmsf(
selection="protein and name C",
xaxis_type= "residue_type_number",
residue_name_format="triple",
label_frequency="all",
highlight_threshold=0.6,
highlight_color="red",
bg_color="white",
fig_bg_color="white",
text_color="black",
show_grid=False,
line_color="#1f77b4",
line_width=2,
#title=" ",
save="trajectory_analysis_example_07.png",
dpi=300,
# other settings...
)
Output figure:
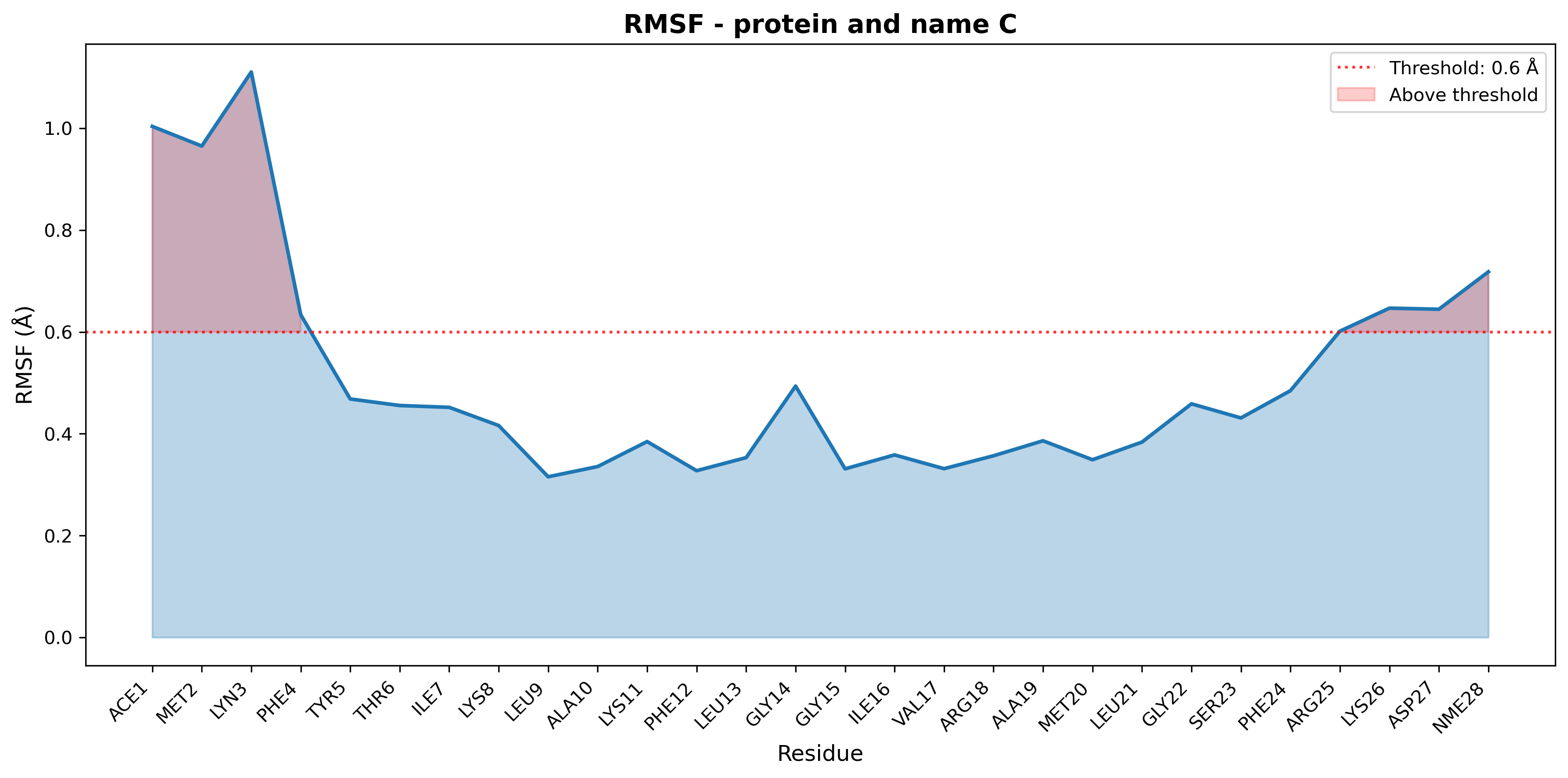
Figure: RMSF plot for multiple with threshold highlight.
Method: calculate_distances()¶
Calculate distances between atom selections over time.
Parameters:
selections(dict): Dictionary of{name: (selection1, selection2)}
Returns: Dictionary with distance data for each named selection:
'time': Time array in nanoseconds'distance': Distance array in Angstroms
Example:
results = analyzer.calculate_distances({
"gate": ("resid 1-3 and name CA", "resid 18-20 and name CA"),
"binding_site": ("resid 100-110", "resname LIG")
})
for name, data in results.items():
print(f"{name}:")
print(f" Time: {data['time'][0]:.3f} - {data['time'][-1]:.3f} ns")
print(f" Distance: {data['distance'].mean():.2f} ± {data['distance'].std():.2f} Å")
Method: plot_distances()¶
Plot distances with full customization including line styling.
plot_distances(
selections: Dict[str, tuple],
distance_units: str = "Å",
time_units: str = "ns",
line_colors: Optional[List[str]] = None,
line_width: float = 1.2,
line_style: str = "-",
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
xlim: Optional[tuple] = None,
ylim: Optional[tuple] = None,
title: Optional[str] = None,
xlabel: Optional[str] = None,
ylabel: Optional[str] = None,
show_mean_lines: bool = True,
hlines: Optional[List[float]] = None,
hline_colors: Optional[List[str]] = None,
hline_styles: Optional[List[str]] = None,
hline_widths: Optional[List[float]] = None,
vlines: Optional[List[float]] = None,
vline_colors: Optional[List[str]] = None,
vline_styles: Optional[List[str]] = None,
vline_widths: Optional[List[float]] = None,
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (10, 6),
dpi: int = 300
)
Parameters:
| Parameter | Type | Default | Description |
|---|---|---|---|
selections | dict | - | Dictionary of {name: (sel1, sel2)} |
distance_units | str | "Å" | Distance units: "Å" or "nm" |
time_units | str | "ns" | Time units: "ps", "ns", or "µs" |
line_colors | list or None | None | List of colors for each distance pair |
line_width | float | 1.2 | Line thickness/width |
line_style | str | "-" | Line style: "-" (solid), "--" (dashed), "-." (dash-dot), ":" (dotted) |
show_mean_lines | bool | True | Show dashed mean distance lines |
hlines | list or None | None | List of Y values for horizontal reference lines |
hline_colors | list or None | None | List of colors for horizontal lines (cycles if fewer than lines) |
hline_styles | list or None | None | List of line styles for horizontal lines (default: "--") |
hline_widths | list or None | None | List of line widths for horizontal lines (default: 1.0) |
vlines | list or None | None | List of X values (time points) for vertical reference lines |
vline_colors | list or None | None | List of colors for vertical lines (cycles if fewer than lines) |
vline_styles | list or None | None | List of line styles for vertical lines (default: "--") |
vline_widths | list or None | None | List of line widths for vertical lines (default: 1.0) |
| ...other params | Same as plot_rmsd() |
Example 8: Distances¶
from pathlib import Path
from gatewizard.utils.namd_analysis import TrajectoryAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Path to topology in equilibration_folder
topology_file = data_dir / "system.pdb"
# Multiple trajectory files from equilibration_folder
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd",
data_dir / "step4_equilibration.dcd",
data_dir / "step5_equilibration.dcd",
data_dir / "step6_equilibration.dcd",
data_dir / "step7_production.dcd",
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = TrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times={
"step1_equilibration.dcd": 0.1, # 100 ps
"step2_equilibration.dcd": 0.1, # 100 ps
"step3_equilibration.dcd": 0.1, # 100 ps
"step4_equilibration.dcd": 0.1, # 100 ps
"step5_equilibration.dcd": 0.1, # 100 ps
"step6_equilibration.dcd": 0.1, # 100 ps
"step7_production.dcd": 0.1 # 100 ps
}
)
# Calculate and plot distances between selections
analyzer.plot_distances(
selections={
"gate_distance": ("resid 1 and name C", "resid 28 and name C"),
"domain_distance": ("resid 1-2 and name C", "resid 9-10 and name C")
},
bg_color="white",
fig_bg_color="white",
text_color="black",
line_width=3,
save="trajectory_analysis_example_08_distances.png"
)
print(f"Distances plot saved: trajectory_analysis_example_08_distances.png")
Output figure:
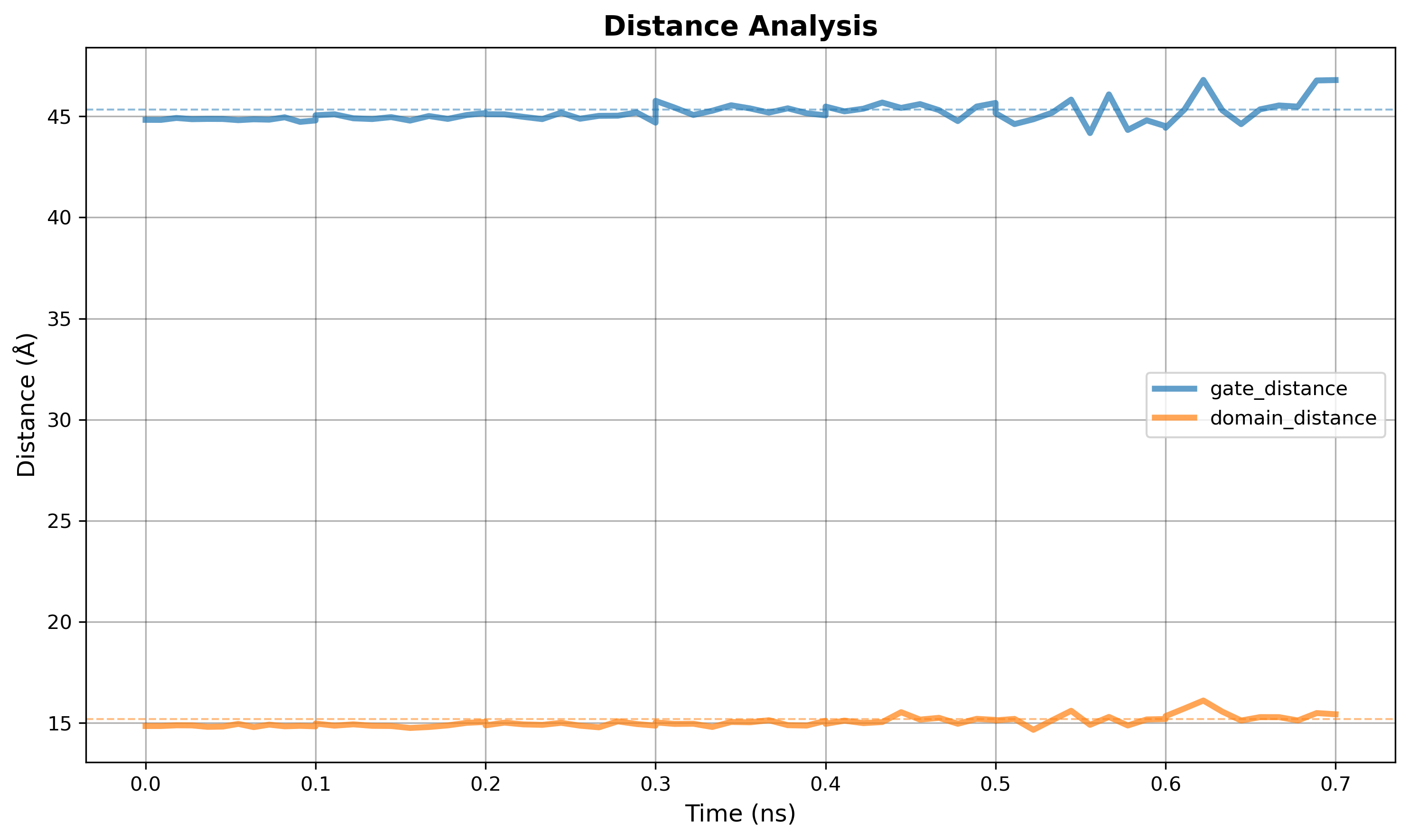
Figure: Distances plot for multiple defined regions on the protein.
Method: calculate_radius_of_gyration()¶
Calculate radius of gyration over trajectory.
Parameters:
selection(str): MDTraj selection string
Returns: Dictionary with:
'time': Time array in nanoseconds'rg': Radius of gyration in Angstroms
Example:
data = analyzer.calculate_radius_of_gyration("protein")
print(f"Rg range: {data['rg'].min():.2f} - {data['rg'].max():.2f} Å")
print(f"Mean Rg: {data['rg'].mean():.2f} Å")
Method: plot_radius_of_gyration()¶
Plot radius of gyration with full customization including line styling and convergence control.
plot_radius_of_gyration(
selection: str = "protein",
distance_units: str = "Å",
time_units: str = "ns",
line_color: str = "purple",
line_width: float = 1.2,
line_style: str = "-",
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
xlim: Optional[tuple] = None,
ylim: Optional[tuple] = None,
title: Optional[str] = None,
xlabel: Optional[str] = None,
ylabel: Optional[str] = None,
show_convergence: bool = True,
convergence_color: str = "red",
convergence_style: str = "--",
convergence_width: float = 1.5,
hlines: Optional[List[float]] = None,
hline_colors: Optional[List[str]] = None,
hline_styles: Optional[List[str]] = None,
hline_widths: Optional[List[float]] = None,
vlines: Optional[List[float]] = None,
vline_colors: Optional[List[str]] = None,
vline_styles: Optional[List[str]] = None,
vline_widths: Optional[List[float]] = None,
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (10, 6),
dpi: int = 300
)
Parameters:
| Parameter | Type | Default | Description |
|---|---|---|---|
selection | str | "protein" | MDTraj selection string |
distance_units | str | "Å" | Distance units: "Å" or "nm" |
time_units | str | "ns" | Time units: "ps", "ns", or "µs" |
line_color | str | "purple" | Plot line color (matplotlib color or hex) |
line_width | float | 1.2 | Line thickness/width |
line_style | str | "-" | Line style: "-" (solid), "--" (dashed), "-." (dash-dot), ":" (dotted) |
show_convergence | bool | True | Show convergence line (mean of last 20% of trajectory) |
convergence_color | str | "red" | Color for convergence line |
convergence_style | str | "--" | Line style for convergence line |
convergence_width | float | 1.5 | Width of convergence line |
hlines | list or None | None | List of Y values for horizontal reference lines |
hline_colors | list or None | None | List of colors for horizontal lines (cycles if fewer than lines) |
hline_styles | list or None | None | List of line styles for horizontal lines (default: "--") |
hline_widths | list or None | None | List of line widths for horizontal lines (default: 1.0) |
vlines | list or None | None | List of X values (time points) for vertical reference lines |
vline_colors | list or None | None | List of colors for vertical lines (cycles if fewer than lines) |
vline_styles | list or None | None | List of line styles for vertical lines (default: "--") |
vline_widths | list or None | None | List of line widths for vertical lines (default: 1.0) |
| ...other params | Same as plot_rmsd() for colors, grid, limits, etc. |
Example 9: Radius of gyration¶
from pathlib import Path
from gatewizard.utils.namd_analysis import TrajectoryAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Path to topology in equilibration_folder
topology_file = data_dir / "system.pdb"
# Multiple trajectory files from equilibration_folder
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd",
data_dir / "step4_equilibration.dcd",
data_dir / "step5_equilibration.dcd",
data_dir / "step6_equilibration.dcd",
data_dir / "step7_production.dcd",
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = TrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times={
"step1_equilibration.dcd": 0.1, # 100 ps
"step2_equilibration.dcd": 0.1, # 100 ps
"step3_equilibration.dcd": 0.1, # 100 ps
"step4_equilibration.dcd": 0.1, # 100 ps
"step5_equilibration.dcd": 0.1, # 100 ps
"step6_equilibration.dcd": 0.1, # 100 ps
"step7_production.dcd": 0.1 # 100 ps
}
)
# Calculate and plot radius of gyration of a selection
analyzer.plot_radius_of_gyration(
selection="protein",
bg_color="white",
fig_bg_color="white",
text_color="black",
line_width=3,
save="trajectory_analysis_example_09_rdgyr.png",
dpi=300,
)
Method: plot_comprehensive()¶
Create a 4-panel analysis plot with RMSD, RMSF, Rg, and statistics.
plot_comprehensive(
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (14, 10),
dpi: int = 300
)
Parameters:
save(str, optional): Filename to save plotshow(bool): Display plot interactivelyfigsize(tuple): Figure sizedpi(int): Resolution
Example:
Tips & Best Practices¶
1. Always Provide file_times for Multiple Files¶
# ✅ GOOD: Proper time scaling
file_times = {
"step1_equilibration.dcd": 0.05, # 50 ps
"step2_equilibration.dcd": 0.05,
"step3_equilibration.dcd": 0.05
}
# ❌ BAD: Frame-based time (incorrect)
file_times = None # Will use frame indices
2. Match Units to Your Analysis¶
# Short simulations (< 1 ns)
time_units="ps", distance_units="Å"
# Medium simulations (1-100 ns)
time_units="ns", distance_units="Å"
# Long simulations (> 100 ns)
time_units="ns", distance_units="nm"
3. RMSF Labeling for Different Protein Sizes¶
# Small protein (< 100 residues)
label_frequency="every_2"
# Medium protein (100-300 residues)
label_frequency="every_5"
# Large protein (> 300 residues)
label_frequency="every_10" or "every_20"
4. Color Schemes¶
# Dark theme (presentations)
bg_color="#2b2b2b", text_color="Auto"
# Light theme (publications)
bg_color="#ffffff", text_color="black"
# Transparent (overlays)
bg_color="none", fig_bg_color="none"
Function: get_equilibration_progress()¶
Get progress information from NAMD equilibration logs.
get_equilibration_progress(
log_directory: Path,
output_file: Optional[Path] = None
) -> Dict[str, NAMDProgress]
Parameters:
log_directory(Path): Directory containing NAMD log filesoutput_file(Path, optional): Save progress summary to JSON
Returns: Dictionary of progress information for each stage
Example:
from gatewizard.utils.namd_analysis import get_equilibration_progress
progress = get_equilibration_progress("equilibration/")
for stage, info in progress.items():
print(f"{stage}: {info.progress_percent:.1f}% complete")
Line Styling and Customization¶
All trajectory analysis plotting methods (plot_rmsd(), plot_rmsf(), plot_distances(), plot_radius_of_gyration()) support line styling options for full control over plot appearance.
Common Line Parameters¶
Available in all plotting methods:
| Parameter | Type | Default | Description |
|---|---|---|---|
line_color | str | varies | Color name (e.g., "blue") or hex code (e.g., "#1f77b4") |
line_width | float | 1.2 | Line thickness (e.g., 0.8, 1.5, 2.5) |
line_style | str | "-" | Line style (see options below) |
Line Style Options¶
| Style | Value | Visual | Use Case |
|---|---|---|---|
| Solid | "-" | ──────── | Default, standard plots |
| Dashed | "--" | ── ── ── | Highlighting, predictions |
| Dash-dot | "-." | ── · ── · | Alternative highlighting |
| Dotted | ":" | · · · · · | Subtle lines, backgrounds |
RMSD & Radius of Gyration Specific¶
Additional convergence line control for plot_rmsd() and plot_radius_of_gyration():
| Parameter | Type | Default | Description |
|---|---|---|---|
show_convergence | bool | True | Toggle convergence line on/off |
convergence_color | str | "red" | Color for convergence line |
convergence_style | str | "--" | Line style for convergence |
convergence_width | float | 1.5 | Width of convergence line |
RMSD Specific¶
Threshold highlighting for plot_rmsd():
| Parameter | Type | Default | Description |
|---|---|---|---|
highlight_threshold | float or None | None | Highlight regions above this RMSD value (in Å or nm) |
RMSF Specific¶
Flexibility highlighting for plot_rmsf():
| Parameter | Type | Default | Description |
|---|---|---|---|
highlight_threshold | float or None | None | Highlight residues above this RMSF value (in Å or nm) |
Quick Reference Examples¶
Thick Dashed Line:
Thin Dotted Line:
Custom Convergence (Green Dash-dot):
No Convergence Line:
RMSD with Threshold:
RMSF with Flexibility Threshold:
MDTraj Selection Syntax¶
Common Selections¶
"protein" # All protein atoms
"protein and backbone" # Backbone atoms (N, CA, C, O)
"protein and name CA" # C-alpha atoms only
"resid 50-150" # Residues 50 to 150
"resid 50-70 and name CA" # C-alpha of residues 50-70
"resname LIG" # Ligand with residue name LIG
"protein and not name H*" # Protein without hydrogens
"within 5 of resname LIG" # Atoms within 5Å of ligand
Combining Selections¶
"(resid 1-50) or (resid 100-150)" # Two regions
"protein and (name CA or name N)" # Multiple atom names
"resid 25 and name NH1 NH2" # Multiple atoms in residue
Class: BilayerTrajectoryAnalyzer¶
Lipid bilayer analysis built on lipyphilic and MDAnalysis.
Constructor¶
BilayerTrajectoryAnalyzer(
topology: Path,
trajectory: Path | List[Path],
file_times: Optional[Dict[str, float]] = None
)
Same topology/trajectory interface as TrajectoryAnalyzer. Leaflet assignment is performed automatically before each analysis.
Method: calculate_area_per_lipid()¶
Calculate the area per lipid via 2D Voronoi tessellation (lipyphilic AreaPerLipid).
calculate_area_per_lipid(
lipid_sel: str = "name PO4",
leaflet_lipid_sel: Optional[str] = None,
start: Optional[int] = None,
stop: Optional[int] = None,
step: Optional[int] = None,
verbose: bool = False,
) -> Dict[str, np.ndarray]
Parameters:
| Parameter | Description |
|---|---|
lipid_sel | Atoms used for Voronoi tessellation. MARTINI: "name GL1 GL2 ROH". All-atom: "name PO4" or phosphate selections. |
leaflet_lipid_sel | Selection for leaflet assignment. Defaults to lipid_sel. |
start, stop, step | Trajectory frame range passed to lipyphilic. |
Returns: time (ns), areas (n_lipids × n_frames, Ų), mean_area_per_lipid, mean_upper_leaflet, mean_lower_leaflet, resids, resnames.
See Example 14: Area per Lipid.
Method: calculate_membrane_thickness()¶
Calculate bilayer thickness from interleaflet headgroup distances (lipyphilic MembThickness).
calculate_membrane_thickness(
lipid_sel: str = "name PO4",
leaflet_lipid_sel: Optional[str] = None,
leaflet_filter_sel: Optional[str] = None,
n_bins: int = 1,
interpolate: bool = False,
start: Optional[int] = None,
stop: Optional[int] = None,
step: Optional[int] = None,
verbose: bool = False,
) -> Dict[str, np.ndarray]
Parameters:
| Parameter | Description |
|---|---|
lipid_sel | Headgroup atoms for the thickness calculation. |
leaflet_lipid_sel | Selection for leaflet assignment. Defaults to lipid_sel. |
leaflet_filter_sel | Optional filter passed to AssignLeaflets.filter_leaflets() to exclude species (e.g. cholesterol). |
n_bins | Grid resolution for intrinsic surface construction (1 = global mean height). |
interpolate | Fill missing grid values when n_bins is large (slower). |
Returns: time (ns), thickness (Å).
See Example 15: Membrane Thickness.
Method: plot_area_per_lipid() / plot_membrane_thickness()¶
Generate time-series plots with the same styling options as TrajectoryAnalyzer plots. See Examples 14 and 15.
Function: run_bilayer_analysis()¶
JSON-serializable API for GUI and downstream consumers.
run_bilayer_analysis(
topology_file: Union[str, Path],
trajectory_files: List[Union[str, Path]],
analysis_type: str,
lipid_sel: str = "name PO4",
leaflet_lipid_sel: Optional[str] = None,
leaflet_filter_sel: Optional[str] = None,
n_bins: int = 1,
interpolate: bool = False,
file_times: Optional[Dict[str, float]] = None,
start: Optional[int] = None,
stop: Optional[int] = None,
step: Optional[int] = None,
verbose: bool = False,
) -> Dict[str, Any]
Supported analysis_type values: area_per_lipid, membrane_thickness (aliases: apl, thickness, memb_thickness).
Area per lipid return shape:
{
"analysis_type": "area_per_lipid",
"x": [...], # time (ns)
"y": [...], # mean area per lipid (Ų)
"mean_upper_leaflet": [...],
"mean_lower_leaflet": [...],
"lipid_resids": [...],
"lipid_resnames": [...],
"per_lipid_areas": [[...], ...],
"stats": {"mean", "std", "min", "max"},
}
Membrane thickness return shape:
{
"analysis_type": "membrane_thickness",
"x": [...],
"y": [...], # thickness (Å)
"n_bins": 1,
"stats": {"mean", "std", "min", "max"},
}
Examples¶
Complete working examples are available in Analysis examples. Each example demonstrates specific analysis capabilities and can be run directly.
Example 10: Dark Theme RMSD Analysis - Full Customization¶
Complete RMSD customization with dark themes showing all available plot options.
from pathlib import Path
from gatewizard.utils.namd_analysis import TrajectoryAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# ============================================================================
# Dark Theme RMSD Analysis - Full Customization
# ============================================================================
topology_file = data_dir / "system.pdb"
# Multiple trajectory files from equilibration_folder
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd",
data_dir / "step4_equilibration.dcd",
]
# Initialize analyzer with custom time for each file (in nanoseconds)
analyzer = TrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times={
"step1_equilibration.dcd": 0.1, # 100 ps
"step2_equilibration.dcd": 0.1, # 100 ps
"step3_equilibration.dcd": 0.1, # 100 ps
"step4_equilibration.dcd": 0.1, # 100 ps
}
)
# RMSD plot with full dark theme customization
analyzer.plot_rmsd(
selection="protein and backbone",
reference_frame=0,
align=True,
distance_units="Å",
time_units="ps",
line_color="#00d9ff", # Bright cyan
line_width=2.0,
line_style="-",
bg_color="#1a1a2e", # Dark blue-black
fig_bg_color="#16213e", # Darker border
text_color="#eee", # Light gray
show_grid=True,
xlim=None,
ylim=None,
title="Dark Theme RMSD - Full Customization",
xlabel="Time (ps)",
ylabel="RMSD (Å)",
highlight_threshold=None,
highlight_color="orange",
highlight_alpha=0.2,
show_convergence=True,
convergence_color="#ff006e", # Magenta
convergence_style="--",
convergence_width=2.0,
hlines=None,
hline_colors=None,
hline_styles=None,
hline_widths=None,
vlines=None,
vline_colors=None,
vline_styles=None,
vline_widths=None,
save="dark_theme_rmsd_example_10.png",
show=False,
figsize=(12, 7),
dpi=300
)
print(f"Dark theme RMSD plot saved: dark_theme_rmsd_example_10.png")
# ============================================================================
# Dark Theme RMSD - With Threshold and Reference Lines
# ============================================================================
analyzer.plot_rmsd(
selection="protein and backbone",
align=True,
distance_units="Å",
time_units="ps",
line_color="#7fff00", # Chartreuse
line_width=1.8,
line_style="-",
bg_color="#0d1117", # GitHub dark
fig_bg_color="#010409",
text_color="#c9d1d9",
show_grid=True,
title="Dark Theme RMSD - With Threshold and Reference Lines",
xlabel="Time (ps)",
ylabel="RMSD (Å)",
highlight_threshold=0.25, # Highlight regions > 0.25 Å
highlight_color="#ff6b6b", # Red highlight
highlight_alpha=0.5,
show_convergence=True,
convergence_color="#ffd700", # Gold
convergence_style="-.",
convergence_width=1.5,
hlines=[0.1, 0.2, 0.25], # Horizontal reference lines
hline_colors=["#4ecdc4", "#95e1d3", "#ff6b6b"], # Teal to red gradient
hline_styles=[":", ":", ":"],
hline_widths=[1.0, 1.0, 1.0],
save="dark_theme_rmsd_with_lines_example_10.png",
figsize=(12, 7),
dpi=300
)
print(f"Dark theme RMSD with lines saved: dark_theme_rmsd_with_lines_example_10.png")
# ============================================================================
# Dark Theme RMSD - Minimal Style
# ============================================================================
analyzer.plot_rmsd(
selection="protein and backbone",
align=True,
distance_units="Å",
time_units="ps",
line_color="#ffffff", # Pure white line
line_width=1.5,
line_style="-",
bg_color="#000000", # Pure black
fig_bg_color="#000000",
text_color="#ffffff",
show_grid=False, # No grid for minimal look
title="", # No title
show_convergence=False, # No convergence line
save="dark_theme_rmsd_minimal_example_10.png",
figsize=(10, 6),
dpi=300
)
print(f"Dark theme RMSD minimal saved: dark_theme_rmsd_minimal_example_10.png")
print(f"\nAll dark theme RMSD figures created with:")
print(f" - Custom dark backgrounds")
print(f" - Full customization options")
print(f" - High resolution (300 DPI)")
Output figures Three dark-themed RMSD plots:
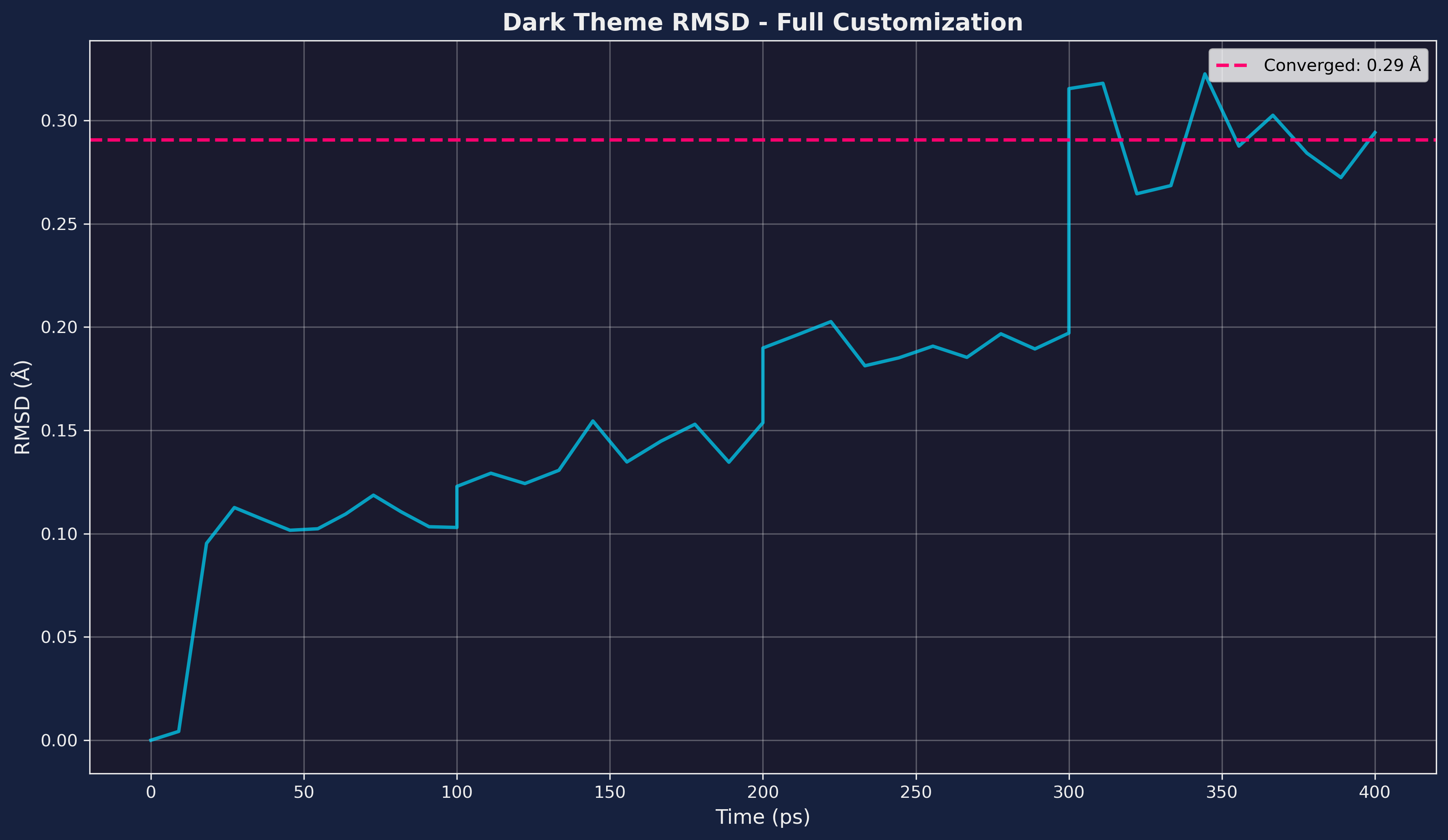
Figure: Dark theme RMSD plot
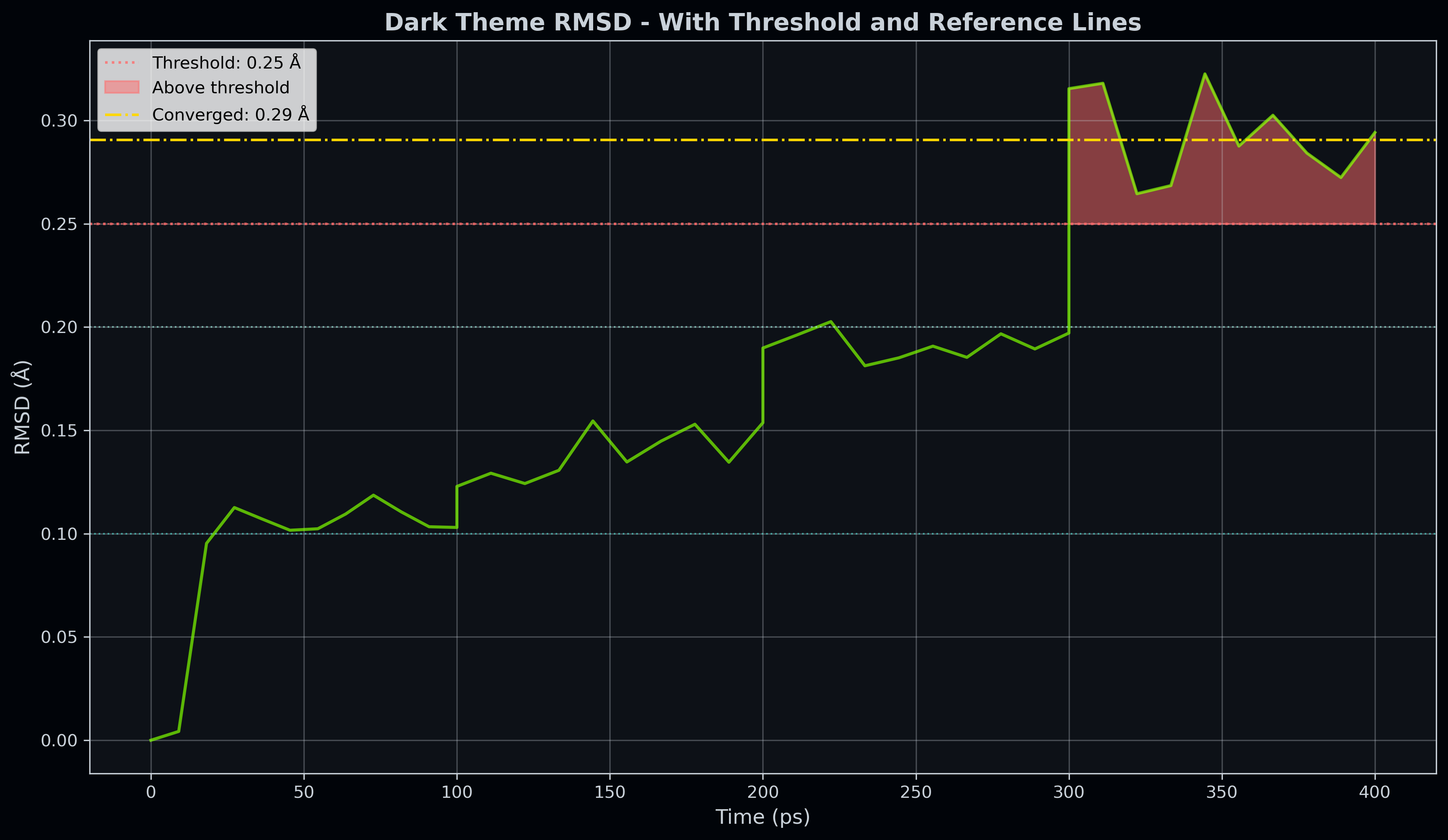
Figure: Dark theme RMSD with lines
Figure: Dark theme RMSD minimal
Class: OpenMMLogAnalyzer¶
Parse and analyze OpenMM StateDataReporter log files. The interface mirrors EnergyAnalyzer so the two can be used interchangeably in analysis workflows.
Constructor
OpenMMLogAnalyzer(
log_file: Union[Path, str, List[Union[Path, str]]],
file_times: Optional[Dict[str, float]] = None,
)
| Parameter | Type | Description |
|---|---|---|
log_file | str \| Path \| list | Path to a single OpenMM log file, or a list of paths for multi-stage analysis (times are concatenated). |
file_times | dict | Optional {filename: duration_ns} mapping that overrides the time axis from the log's "Time (ps)" column. Useful when logs lack the time column or have restarted counters. |
Key differences from EnergyAnalyzer:
| Feature | EnergyAnalyzer (NAMD) | OpenMMLogAnalyzer (OpenMM) |
|---|---|---|
| Native energy units | kcal/mol | kJ/mol |
Color kwarg in plot_properties | line_colors | colors |
| Log format | space-delimited .log | tab-delimited (StateDataReporter) |
Available property keys (depend on what StateDataReporter reports):
| Key | Description |
|---|---|
potential | Potential energy (kJ/mol) |
kinetic | Kinetic energy (kJ/mol) |
total | Total energy (kJ/mol) |
temp | Temperature (K) |
volume | Box volume (nm³) |
density | Density (g/mL) |
Method: get_statistics()¶
Returns mean / std / min / max / initial / final for every numeric column that contains data.
Returns {key: {"mean", "std", "min", "max", "initial", "final"}}
Method: plot_energy()¶
analyzer.plot_energy(
energy_units: str = "kJ/mol",
time_units: str = "ns",
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
title: Optional[str] = None,
target_temperature: Optional[float] = None,
target_pressure: Optional[float] = None,
save: Optional[str] = None,
show: bool = False,
figsize: tuple = (12, 10),
dpi: int = 300,
)
2×2 energy summary plot — total energy, potential + kinetic, temperature, volume / density.
| Parameter | Default | Description |
|---|---|---|
energy_units | 'kJ/mol' | 'kJ/mol' or 'kcal/mol' |
time_units | 'ns' | 'ps', 'ns', or 'µs' |
target_temperature | None | Reference temperature (K) drawn as a horizontal dashed line |
save | None | Filename for saved figure |
show | False | Display interactively |
Method: plot_properties()¶
analyzer.plot_properties(
properties: Optional[List[str]] = None,
energy_units: str = "kJ/mol",
time_units: str = "ns",
bg_color: str = "#2b2b2b",
fig_bg_color: str = "#212121",
text_color: str = "Auto",
show_grid: bool = True,
separate_plots: bool = False,
save: Optional[str] = None,
show: bool = False,
figsize: Optional[tuple] = None,
dpi: int = 300,
xlim: Optional[tuple] = None,
ylim: Optional[tuple] = None,
colors: Optional[List[str]] = None,
)
Plot selected properties vs time. If properties is not provided, all non-empty numeric columns are plotted automatically.
| Parameter | Default | Description |
|---|---|---|
properties | None (all) | List of keys to plot, e.g. ["potential", "temp", "volume"] |
energy_units | 'kJ/mol' | 'kJ/mol' or 'kcal/mol' |
separate_plots | False | Save/show each property as its own figure |
colors | None | Line color list (one per property). Unlike EnergyAnalyzer, the kwarg is colors, not line_colors. |
save | None | Filename (or prefix when separate_plots=True) |
show | False | Display interactively |
Example 11: Basic Energy Analysis¶
from pathlib import Path
from gatewizard.utils.openmm_analysis import OpenMMLogAnalyzer
# Get the directory where this script is located
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Single OpenMM log file (StateDataReporter output)
log_file = data_dir / "step7_production.log"
# Initialize the analyzer
analyzer = OpenMMLogAnalyzer(log_file)
# ------------------------------------------------------------------
# 1. Get statistics for all properties
# ------------------------------------------------------------------
stats = analyzer.get_statistics()
for key, s in stats.items():
print(
f" {key:20s} mean={s['mean']:12.3f} std={s['std']:10.3f}"
f" min={s['min']:12.3f} max={s['max']:12.3f}"
)
# → All values in kJ/mol (energies) or native units (temperature in K,
# volume in nm³, density in g/mL)
# ------------------------------------------------------------------
# 2. 2×2 summary plot: total energy, potential + kinetic, temperature,
# volume / density
# ------------------------------------------------------------------
analyzer.plot_energy(
save="energy_summary_example_11.png",
show=False,
target_temperature=303.15,
)
print("Energy summary saved: energy_summary_example_11.png")
# ------------------------------------------------------------------
# 3. Same plot converted to kcal/mol
# ------------------------------------------------------------------
analyzer.plot_energy(
energy_units="kcal/mol",
save="energy_summary_kcal_example_11.png",
show=False,
)
print("Energy summary (kcal/mol) saved: energy_summary_kcal_example_11.png")
Example 12: Multi-Stage Analysis¶
from pathlib import Path
from gatewizard.utils.openmm_analysis import OpenMMLogAnalyzer
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
# Pass a list of log files — time axis is concatenated automatically
log_files = [
data_dir / "step1_equilibration.log",
data_dir / "step2_equilibration.log",
data_dir / "step3_equilibration.log",
data_dir / "step4_equilibration.log",
data_dir / "step5_equilibration.log",
data_dir / "step6_equilibration.log",
data_dir / "step7_production.log",
]
# Provide real stage durations (ns) to override the "Time (ps)" column
# — useful when logs lack the time column or have restarted counters.
file_times = {
"step1_equilibration.log": 0.125,
"step2_equilibration.log": 0.125,
"step3_equilibration.log": 0.125,
"step4_equilibration.log": 0.25,
"step5_equilibration.log": 0.25,
"step6_equilibration.log": 0.5,
"step7_production.log": 50.0,
}
analyzer = OpenMMLogAnalyzer(log_files, file_times=file_times)
# ------------------------------------------------------------------
# 1. Print key statistics
# ------------------------------------------------------------------
stats = analyzer.get_statistics()
print("=== Multi-stage OpenMM analysis ===")
for key in ("potential", "kinetic", "total", "temp", "volume", "density"):
if key not in stats:
continue
s = stats[key]
print(
f" {key:12s} mean={s['mean']:12.3f}"
f" initial={s['initial']:12.3f} final={s['final']:12.3f}"
)
# ------------------------------------------------------------------
# 2. Energy summary over the full trajectory
# ------------------------------------------------------------------
analyzer.plot_energy(
save="energy_multistage_example_12.png",
show=False,
title="Full equilibration + production (51.375 ns)",
target_temperature=303.15,
)
print("Saved: energy_multistage_example_12.png")
Example 13: Custom Property Plots¶
from pathlib import Path
from gatewizard.utils.openmm_analysis import OpenMMLogAnalyzer
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
log_file = data_dir / "step7_production.log"
analyzer = OpenMMLogAnalyzer(log_file)
# ------------------------------------------------------------------
# 1. Plot all available properties (auto-detected)
# ------------------------------------------------------------------
analyzer.plot_properties(
save="all_properties_example_13.png",
show=False,
)
print("All-properties plot saved: all_properties_example_13.png")
# ------------------------------------------------------------------
# 2. Plot only energies, converted to kcal/mol
# ------------------------------------------------------------------
analyzer.plot_properties(
properties=["potential", "kinetic", "total"],
energy_units="kcal/mol",
save="energies_kcal_example_13.png",
show=False,
colors=["#61afef", "#98c379", "#e06c75"],
)
print("Energy plots (kcal/mol) saved: energies_kcal_example_13.png")
# ------------------------------------------------------------------
# 3. Temperature and density as separate figures
# ------------------------------------------------------------------
analyzer.plot_properties(
properties=["temp", "density"],
separate_plots=True,
save="temp_density_example_13",
show=False,
)
print("Separate figures saved: temp_density_example_13_temp.png, temp_density_example_13_density.png")
Example 14: Area per Lipid¶
Calculate and plot the area per lipid using lipyphilic Voronoi tessellation on the membrane-protein equilibration trajectories in equilibration_folder.
from pathlib import Path
from gatewizard.utils.namd_analysis import BilayerTrajectoryAnalyzer, run_bilayer_analysis
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
topology_file = data_dir / "system.pdb"
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd",
]
# POPC headgroup phosphorus atoms in the AMBER lipid parametrization
LIPID_SEL = "resname PC and name P31"
file_times = {
"step1_equilibration.dcd": 0.1, # 0.1 ns
"step2_equilibration.dcd": 0.1, # 0.1 ns
"step3_equilibration.dcd": 0.1, # 0.1 ns
}
# ------------------------------------------------------------------
# 1. Class API — calculate area per lipid
# ------------------------------------------------------------------
analyzer = BilayerTrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times=file_times,
)
data = analyzer.calculate_area_per_lipid(lipid_sel=LIPID_SEL)
mean_area = float(data["mean_area_per_lipid"].mean())
print(f"Mean area per lipid: {mean_area:.1f} Ų")
print(f"Lipids analysed: {len(data['resids'])}")
print(f"Frames analysed: {data['areas'].shape[1]}")
# ------------------------------------------------------------------
# 2. Plot mean area per lipid time series
# ------------------------------------------------------------------
analyzer.plot_area_per_lipid(
lipid_sel=LIPID_SEL,
series="mean",
time_units="ns",
save="area_per_lipid_example_14.png",
show=False,
)
print("Plot saved: area_per_lipid_example_14.png")
# ------------------------------------------------------------------
# 3. JSON API — run_bilayer_analysis()
# ------------------------------------------------------------------
result = run_bilayer_analysis(
topology_file,
trajectory_files,
analysis_type="area_per_lipid",
lipid_sel=LIPID_SEL,
file_times=file_times,
)
print(f"JSON API mean area: {result['stats']['mean']:.1f} Ų")
For other force fields, adjust lipid_sel to match your headgroup atoms, e.g. "name GL1 GL2 ROH" (MARTINI) or "name PO4" (all-atom CHARMM).
Example 15: Membrane Thickness¶
Calculate and plot bilayer thickness as the mean interleaflet headgroup distance (lipyphilic MembThickness) on the same equilibration trajectories.
from pathlib import Path
from gatewizard.utils.namd_analysis import BilayerTrajectoryAnalyzer, run_bilayer_analysis
script_dir = Path(__file__).parent
data_dir = script_dir / "equilibration_folder"
topology_file = data_dir / "system.pdb"
trajectory_files = [
data_dir / "step1_equilibration.dcd",
data_dir / "step2_equilibration.dcd",
data_dir / "step3_equilibration.dcd",
]
LIPID_SEL = "resname PC and name P31"
file_times = {
"step1_equilibration.dcd": 0.1, # 0.1 ns
"step2_equilibration.dcd": 0.1, # 0.1 ns
"step3_equilibration.dcd": 0.1, # 0.1 ns
}
# ------------------------------------------------------------------
# 1. Class API — calculate membrane thickness
# ------------------------------------------------------------------
analyzer = BilayerTrajectoryAnalyzer(
topology_file,
trajectory_files,
file_times=file_times,
)
data = analyzer.calculate_membrane_thickness(lipid_sel=LIPID_SEL)
mean_thickness = float(data["thickness"].mean())
print(f"Mean membrane thickness: {mean_thickness:.1f} Å")
print(f"Frames analysed: {len(data['thickness'])}")
# ------------------------------------------------------------------
# 2. Plot membrane thickness time series
# ------------------------------------------------------------------
analyzer.plot_membrane_thickness(
lipid_sel=LIPID_SEL,
time_units="ns",
save="membrane_thickness_example_15.png",
show=False,
)
print("Plot saved: membrane_thickness_example_15.png")
# ------------------------------------------------------------------
# 3. JSON API — run_bilayer_analysis()
# ------------------------------------------------------------------
result = run_bilayer_analysis(
topology_file,
trajectory_files,
analysis_type="membrane_thickness",
lipid_sel=LIPID_SEL,
file_times=file_times,
)
print(f"JSON API mean thickness: {result['stats']['mean']:.1f} Å")
To exclude cholesterol from a mixed bilayer thickness calculation, pass leaflet_filter_sel="resname DPPC DOPC" and set lipid_sel to the headgroup atoms of the lipids included in the calculation.
See Also¶
- User Guide - Complete usage guide
- Examples - Working code examples
- Troubleshooting - Common issues